Fe4N2 - 1 - system construction with AVAS and CASSCF
In this tutorial, which serves as the introductory part of a three-part series (second part and third part), we will explore the classical workflow that must be followed before moving on to quantum computations. We will focus on a system that is more complex compared to a simple diatomic molecule like H\(_2\). Additional details regarding the chemical systems, methodologies, and outcomes presented in this tutorial series are available in the associated research paper.
The first step is to run Hartree-Fock (HF) calculations and in our case restricted open-shell Hartree-Fock (ROHF) with the Los Alamos National Laboratory (lanl2dz) effective core potential (ecp) and basis set. We use an InQuanto-PySCF driver to perform the molecular ROHF calculations, and store the results in an InQuanto QubitOperator.
The geometry of Fe\(_4\)N\(_2\) has been optimized for several possible spin multiplicities (7, 9, 11 and 13) and we have found that the state with multiplicity (2S+1) 7 has the lowest energy.
Parameters used:
geometry– Molecular geometry.basis– Atomic basis set valid for Mole class.ecp– Effective core potentials.charge– Total charge.multiplicity– Spin multiplicity, 2S+1.point_group_symmetry– Enable point group symmetry.soscf– Use Second-Order SCF solver (Newton’s method).verbose– Control PySCF verbosity.
Even with a very good initial guess, making the Self-consistent field (SCF) procedure converge is sometimes challenging. To achieve quadratic convergence in the orbital optimization, PySCF implements a general second-order solver called the co-iterative augmented hessian (CIAH) method. By setting the soscf parameter to True, you can activate this method.
[ ]:
%reset -f
# native pyscf
from pyscf import gto, scf
geometry = [
["Fe",[ 0.27291543, -1.18421072, -0.99978567]],
["Fe",[ -1.35922190, 0.45608027, -0.52389493]],
["Fe",[ -0.97901234, -1.08834609, 1.07468517]],
["Fe",[ 0.30675371, 0.72527495, 1.22446007]],
["N", [ 1.32881018, 0.05991762, -0.18585483]],
["N", [ 0.42975492, 1.03128398, -0.58960981]],
]
basis = "lanl2dz"
ecp = {'Fe':'lanl2dz'}
point_group_symmetry = False
multiplicity = 7
charge = 0
verbose = 1
import warnings
warnings.filterwarnings('ignore')
[ ]:
from inquanto.extensions.pyscf import ChemistryDriverPySCFMolecularROHF, CASSCF
driver = ChemistryDriverPySCFMolecularROHF(geometry=geometry, basis=basis, ecp=ecp,charge=charge,
multiplicity=multiplicity, point_group_symmetry=point_group_symmetry,
soscf=True, verbose=verbose)
driver.run_hf()
-598.4570485072064
The NGLView visualizer for InQuanto can be used to display the system.
[ ]:
from inquanto.geometries import GeometryMolecular
from inquanto.extensions.nglview import VisualizerNGL
fe4n2_geom = GeometryMolecular(geometry)
visualizer = VisualizerNGL(fe4n2_geom)
[ ]:
#visualizer.visualize_molecule(atom_labels="index")
To limit the hardware resources we can employ a smaller active space using Atomic Valence Active Space (AVAS) approximation in the PySCF extension of InQuanto. AVAS is a simple and well-defined automated technique for constructing active orbital spaces for use in multi-configuration and multireference electronic structure calculations. Concretely, the technique constructs active molecular orbitals capable of describing all relevant electronic configurations emerging from a targeted set of atomic valence orbitals (e.g., the metal d orbitals in a coordination complex). This is achieved via a linear transformation of the occupied and unoccupied orbital spaces from an easily obtainable single-reference wavefunction (such as from a Hartree-Fock or Kohn-Sham calculations) based on projectors to targeted atomic valence orbitals. More information can be found in the original paper.
Parameters used:
aolabels– AO labels for AO active space.aolabels_vir– If given, separate AO labels for the virtual orbitals. If not given, aolabels is used.n_occ– None or number of localized occupied orbitals to create. If specified, the value of threshold is ignored.n_vir– None or number of localized virtual orbitals to create. If specified, the value of threshold_vir is ignored.force_halves_active– How to handle singly-occupied orbitals in the active space. The singly-occupied orbitals are projected as part of alpha orbitals if False (default), or completely kept in active space if True.freeze_half_filled– If True, all half-filled orbitals (if present) are frozen, i.e. excluded from the AVAS transformation and from the active space.
The reference AO basis for the virtual orbitals is always the computational basis, in contrast to original AVAS but as in the RE method.
For the Fe\(_4\)N\(_2\) cluster if one constructs the active space from (partially) filled Fe d orbitals and the N-N occupied and virtual orbitals, no excitations from Fe 3d to N-N \(\sigma\) or \(\pi\) orbitals are found in the CI wavefunction. We have also separately verified that excitations from the occupied N-N orbitals to the empty Fe-3d orbitals do not contribute. This means that the excitations in the Fe d manifold and in the N-N bond are not coupled and that a reduced active space can be created consisting of orbitals of the dinitrogen system and selected Fe orbitals interacting with it, i.e. excluding all half-filled 3d-like orbitals on Fe.
Here, we directly specify the number of localized occupied orbitals (n_occ) and the number of localized virtual orbitals (n_vir). Our selection of the occupied and virtual orbitals is influenced by the available computational resources. In this particular problem, we opt for a compact (4,3) active space, which is well-suited for the upcoming quantum calculations involving 6 qubits.
Alternatively, it is also possible to define the values of threshold and threshold_vir. These represent the truncation thresholds for the AO-projector, above which atomic orbitals (AOs) are retained within the active space for occupied and virtual orbitals, respectively.
[ ]:
from inquanto.extensions.pyscf import AVAS
ao_pattern = ['N 2p']
ao_pattern_vir = ['N 2p', 'Fe 3d']
avas = AVAS(aolabels=ao_pattern, aolabels_vir=ao_pattern_vir,
n_occ=2, n_vir=1,force_halves_active=False,
freeze_half_filled=True, verbose=verbose)
We can now create a fresh InQuanto Hamiltonian object using the active space we have defined with AVAS.
Parameters used:
frozen– Frozen orbital information.transf– Orbital transformer.
[ ]:
driver_AVAS = ChemistryDriverPySCFMolecularROHF(geometry=geometry, basis=basis, ecp=ecp,charge=charge,
multiplicity=multiplicity, point_group_symmetry=point_group_symmetry,
soscf=True, transf=avas, frozen=avas.frozenf, verbose=verbose)
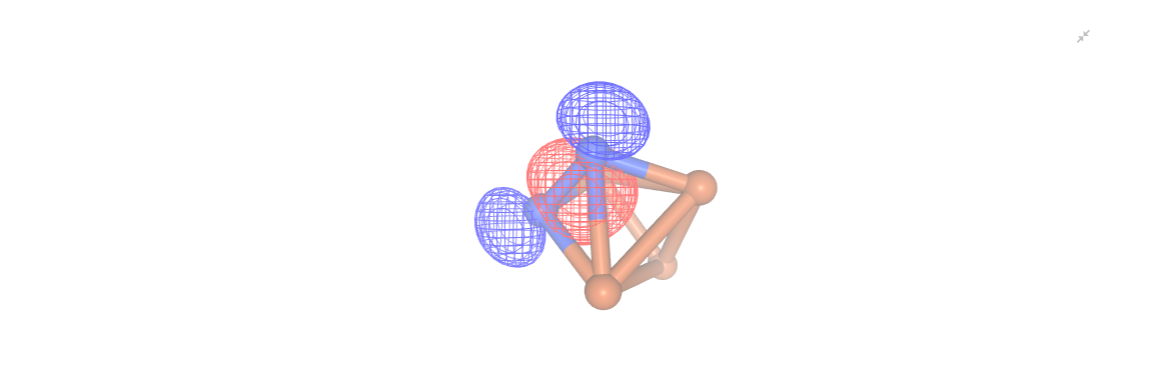
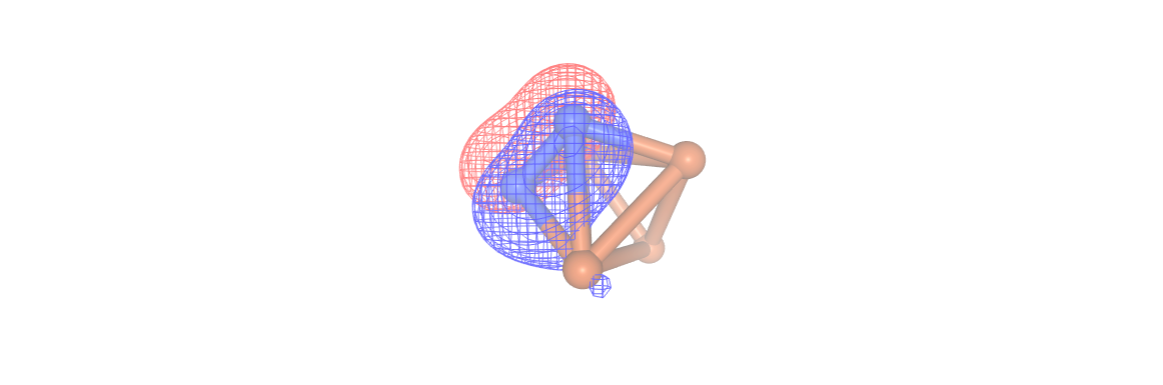
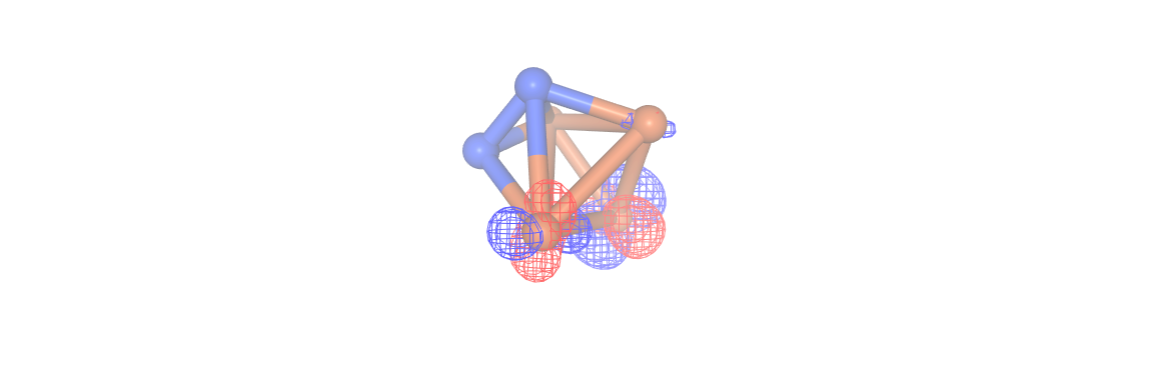
The next step involves performing CASSCF calculations. However, as these calculations tend to be more computationally intensive, it is prudent to visualize the selected active orbitals beforehand. Once more, we will utilize the NGLView visualizer for this purpose.
[ ]:
orbital_cubes = driver_AVAS.get_cube_orbitals()
orbitals = [visualizer.visualize_orbitals(orb,red_isolevel=-2.0, blue_isolevel=2.0) for orb in orbital_cubes]
[ ]:
#orbitals[0]
[ ]:
#orbitals[1]
[ ]:
#orbitals[2]
For the CASSCF calculation, we will employ the most minimal strategy, which involves selecting orbitals (and electrons) around the Fermi level to match the user-specified number of orbitals and electrons. More information on running CASSCF with InQuanto-PySCF extension can be found here.
In order to construct the Hamiltonian based on the CASSCF orbitals, it is necessary to create a new, final InQuanto Hamiltonian object. In this instance, we have employed 9 active spatial orbitals and 10 active electrons for the CASSCF calculation. This choice strikes a balance between obtaining an accurate representation of our system and conducting a calculation that does not demand excessive computational resources.
Additionally, in this step we can analyze the configuration interaction (CI) coefficients by setting the print_ci_coeff parameter to True. This is very important in deciding the suitability of our chosen active space. Notably, only the first[0], second[1], and ninth[8] active orbitals are significantly active. We can visualize these orbitals using NGLView visualizer. It is important to avoid a scenario where only the d orbitals of the Fe atoms are active, as this would result in a state
comprising an equal mixture of 6 configurations distinguished by the placement of electrons within the d orbitals. Expanding the active space to include the orbitals of the nitrogen atoms would lead to an overly large active space, involving a minimum of 10 spatial orbitals.
There is not any standardized approach for selecting the atomic orbital (AO) labels and the number of localized occupied and virtual orbitals. However, the choice depends on the system’s characteristics and the allowable size of the active space. We can manipulate various attributes of the AVAS method to achieve the desired outcome, adapting it to the specific needs of the system.
Parameters used:
init_orbitals– Initial orbital coefficients, can be from a different geometry.print_ci_coeff– If True, the determinants which have coefficients > ci_print_cutoff will be printed out.ci_print_cutoff– Tolerance for printing CI coefficients.max_cycle– Maximum number of CASSCF macroiterations
[ ]:
import numpy as np
final_driver = ChemistryDriverPySCFMolecularROHF(geometry=geometry, basis=basis, ecp=ecp,charge=charge,
multiplicity=multiplicity, point_group_symmetry=point_group_symmetry,
soscf=True, verbose=verbose, frozen=avas.frozenf,
transf=CASSCF(9, 10, init_orbitals=driver_AVAS.get_orbital_coefficients(),
print_ci_coeff=True, ci_print_cutoff=0.03, max_cycle=100))
hamiltonian, space, state = final_driver.get_system()
qubit_hamiltonian = hamiltonian.qubit_encode()
** Largest CI components **
[alpha occ-orbitals] [beta occ-orbitals] CI coefficient
[0 1 2 3 4 5 6 7] [0 1] -0.914034639529
[0 1 2 3 4 5 6 7] [0 8] -0.087749343782
[0 1 2 4 5 6 7 8] [0 3] -0.109127674188
[0 1 3 4 5 6 7 8] [0 1] -0.033936544018
[0 1 3 4 5 6 7 8] [0 8] 0.039567732683
[0 2 3 4 5 6 7 8] [0 1] 0.089215434900
[0 2 3 4 5 6 7 8] [0 2] 0.083166002823
[0 2 3 4 5 6 7 8] [0 8] 0.342939298962
[1 2 3 4 5 6 7 8] [1 8] 0.054090558795
Once more, we employ the NGLView visualizer to display the orbitals using our final InQuanto driver.
[ ]:
orbital_cubes_final = final_driver.get_cube_orbitals()
orbitals_final = [visualizer.visualize_orbitals(orb,red_isolevel=-2.0, blue_isolevel=2.0) for orb in orbital_cubes_final]
[ ]:
#orbitals_final[0]
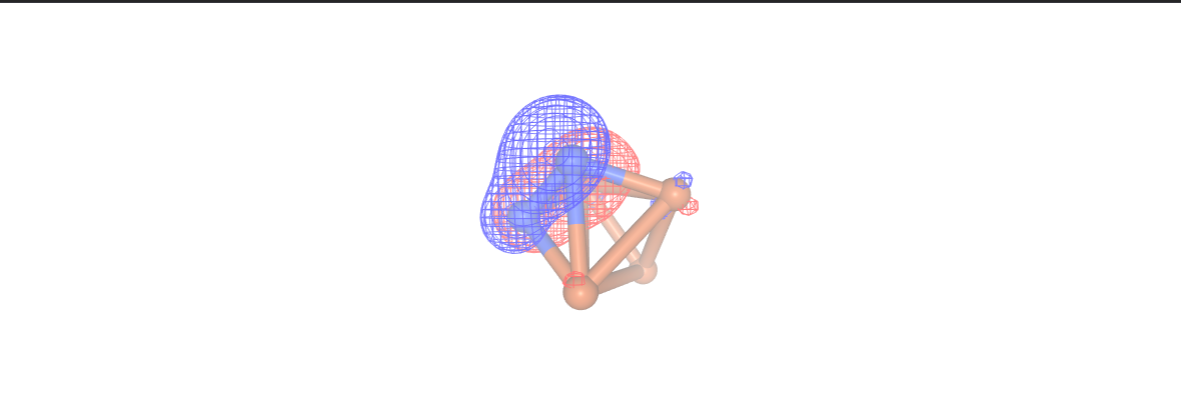
[ ]:
#orbitals_final[1]
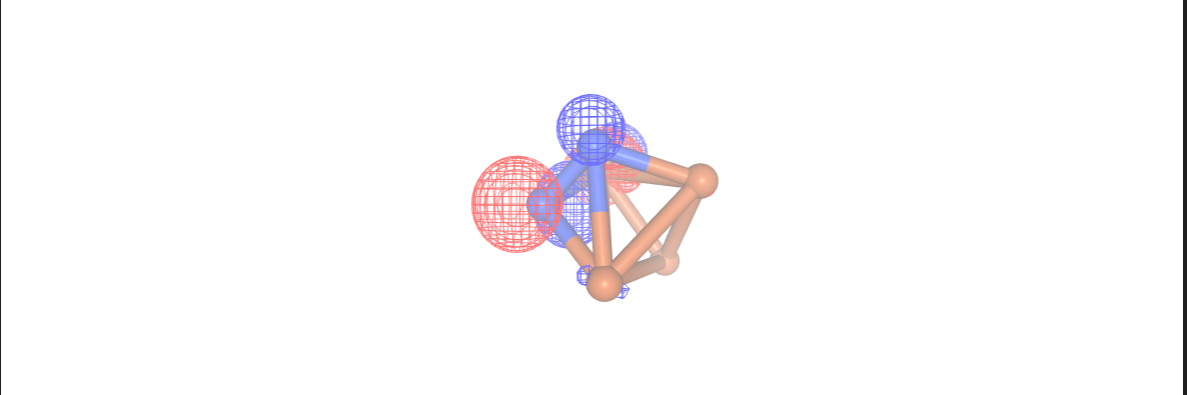
[ ]:
#orbitals_final[2]
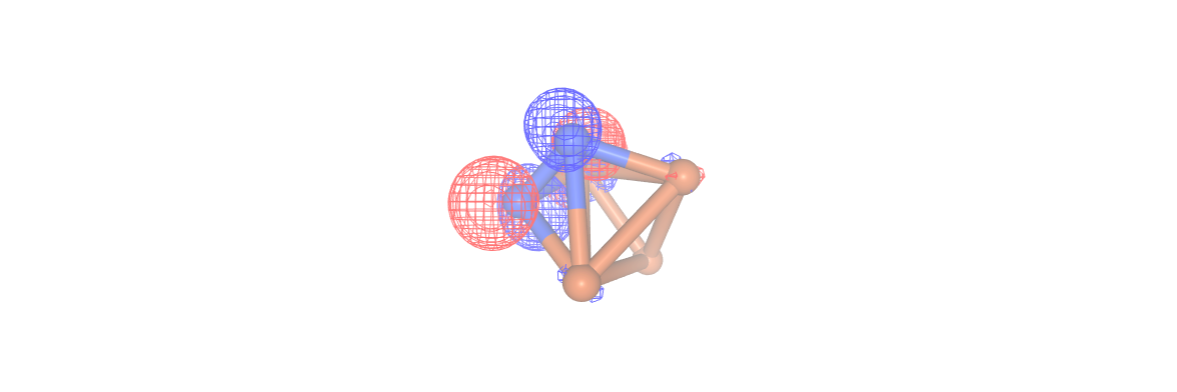
Once the space and state are defined, we can utilize the print_state method to showcase the fermionic state and verify the number of qubits required for the quantum calculations.
[ ]:
space.print_state(state)
0 0a : 1
1 0b : 1
2 1a : 1
3 1b : 1
4 2a : 0
5 2b : 0
As a final step, we can pickle the qubit Hamiltonian, space and state making it convenient for us to import and utilize them in the second part of this tutorial.
[ ]:
import pickle
with open('InQ_tut_fe4n2_space.pickle', 'wb') as handle:
pickle.dump(space, handle, protocol=pickle.HIGHEST_PROTOCOL)
with open('InQ_tut_fe4n2_state.pickle', 'wb') as handle:
pickle.dump(state, handle, protocol=pickle.HIGHEST_PROTOCOL)
with open('InQ_tut_fe4n2_qubit_hamiltonian.pickle', 'wb') as handle:
pickle.dump(qubit_hamiltonian, handle, protocol=pickle.HIGHEST_PROTOCOL)